Mendelian Randomization Workflow
mr-workflow.RmdEnd-to-End MR Analysis
This vignette demonstrates a complete workflow for Mendelian Randomization analysis.
1. Data Preparation
We’ll start by creating mock GWAS data for both an exposure and an outcome.
set.seed(123)
# Mock exposure data
exp_raw <- data.frame(
SNP = paste0("rs", 1:30),
beta = rnorm(30, 0.5, 0.1),
se = runif(30, 0.01, 0.05),
effect_allele = "A",
other_allele = "G",
eaf = runif(30, 0.1, 0.9),
pval = 10^(-runif(30, 5, 10)),
samplesize = 10000,
phenotype = "Exposure Trait"
)
# Mock outcome data
out_raw <- data.frame(
SNP = paste0("rs", 1:100),
beta = rnorm(100, 0, 0.1),
se = runif(100, 0.02, 0.06),
effect_allele = "A",
other_allele = "G",
eaf = runif(100, 0.1, 0.9),
pval = runif(100),
phenotype = "Outcome Trait"
)
exp_dat <- format_data(exp_raw, type = "exposure")
out_dat <- format_data(out_raw, type = "outcome")2. Instrument Strength
The functions package provides utilities to calculate
instrument strength
(
and
-statistic).
Variance Explained ()
Calculate the proportion of variance explained by the instruments
using calculate_r2.
exp_dat <- calculate_r2(exp_dat)
head(exp_dat |> select(SNP, beta.exposure, r2))
#> SNP beta.exposure r2
#> 1 rs1 0.4439524 0.01449638
#> 2 rs2 0.4769823 0.10680549
#> 3 rs3 0.6558708 0.06269876
#> 4 rs4 0.5070508 0.05521028
#> 5 rs5 0.5129288 0.01429991
#> 6 rs6 0.6715065 0.05460598F-statistic from
The F-statistic can be calculated based on , sample size (), and number of instruments ().
# grouping_column determines how to aggregate SNPs (e.g., by phenotype/exposure)
f_stat_res <- calculate_fstat_from_r2(exp_dat, grouping_column = exposure)
unique(f_stat_res |> select(exposure, mean_fstat_from_r2))
#> # A tibble: 1 × 2
#> exposure mean_fstat_from_r2
#> <chr> <dbl>
#> 1 exposure 16.1F-statistic from Beta and SE
Alternatively, calculate the F-statistic directly from effect sizes and standard errors.
exp_dat <- calculate_fstat_from_beta_se(exp_dat)
head(exp_dat |> select(SNP, fstat_from_b_se, mean_f_stats))
#> SNP fstat_from_b_se mean_f_stats
#> 1 rs1 147.0962 484.5334
#> 2 rs2 1195.7696 484.5334
#> 3 rs3 668.9286 484.5334
#> 4 rs4 584.3657 484.5334
#> 5 rs5 145.0737 484.5334
#> 6 rs6 577.6002 484.53343. Harmonization and Analysis
harmonized <- TwoSampleMR::harmonise_data(exp_dat, out_dat, action = 2)
res <- TwoSampleMR::mr(harmonized)4. Custom Visualization
The functions package provides enhanced versions of
standard MR plots. Key differences from the TwoSampleMR
package include: - Improved Labelling: Plots
automatically include trait names in titles and axes. - SNP
Ordering: Forest and Leave-one-out plots order SNPs by effect
size for better clarity. - Styling: Uses custom color
palettes and unified themes suitable for publication.
Scatter Plot
Enhances the standard scatter plot by adding trait labels and enforcing positive exposure effects (flipping signs for consistency).
# Returns a list of plots
plots <- mr_scatter_plot(res, harmonized)
plots[[1]]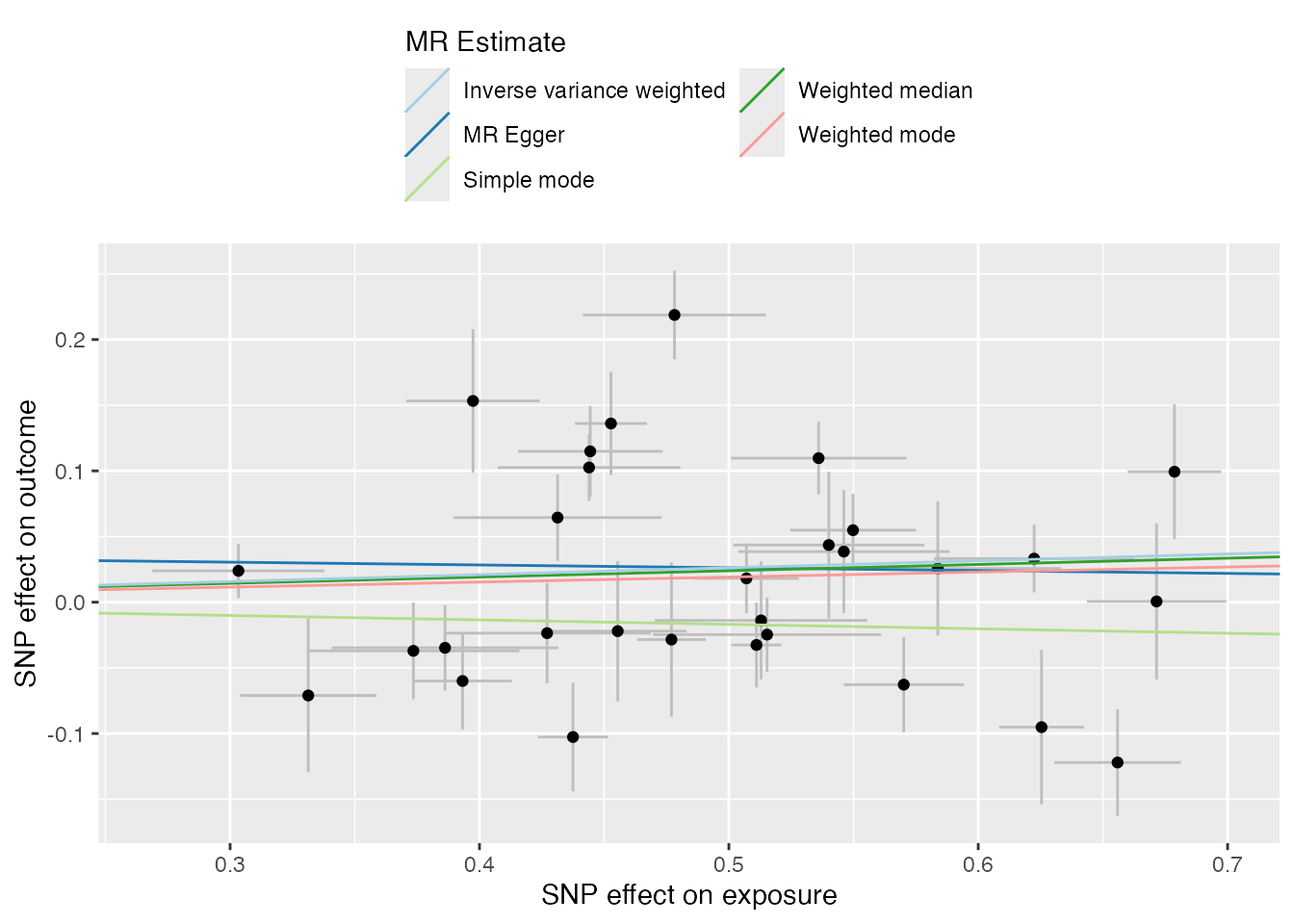
Forest Plot
Standardizes method names (e.g., “IVW”) and separates individual SNP effects from pooled estimates with a gap.
singlesnp_res <- TwoSampleMR::mr_singlesnp(harmonized)
plots <- mr_forest_plot(singlesnp_res)
plots[[1]]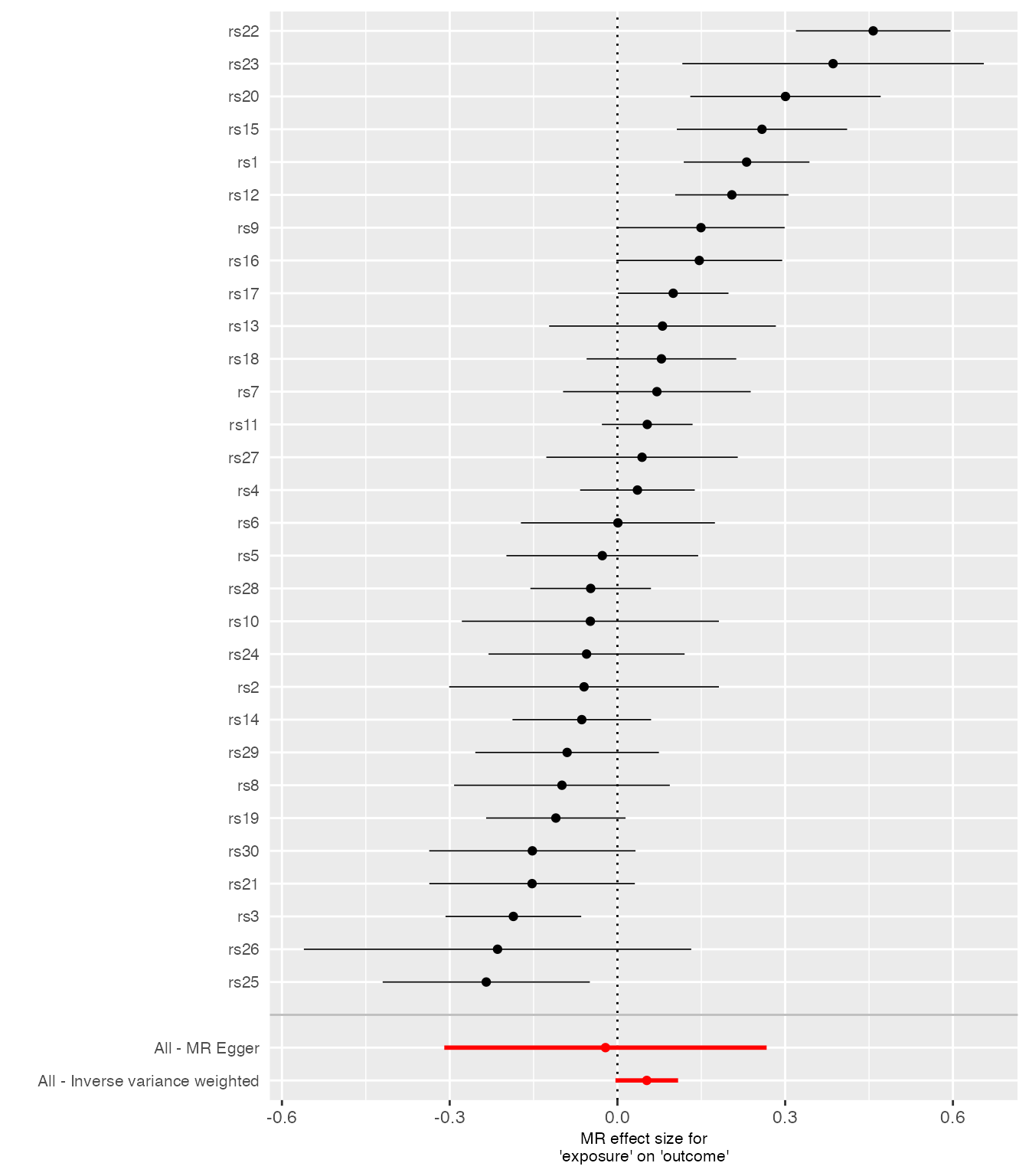
Funnel Plot
Uses a specific high-contrast color palette and clear title mapping.
plots <- mr_funnel_plot(singlesnp_res)
plots[[1]]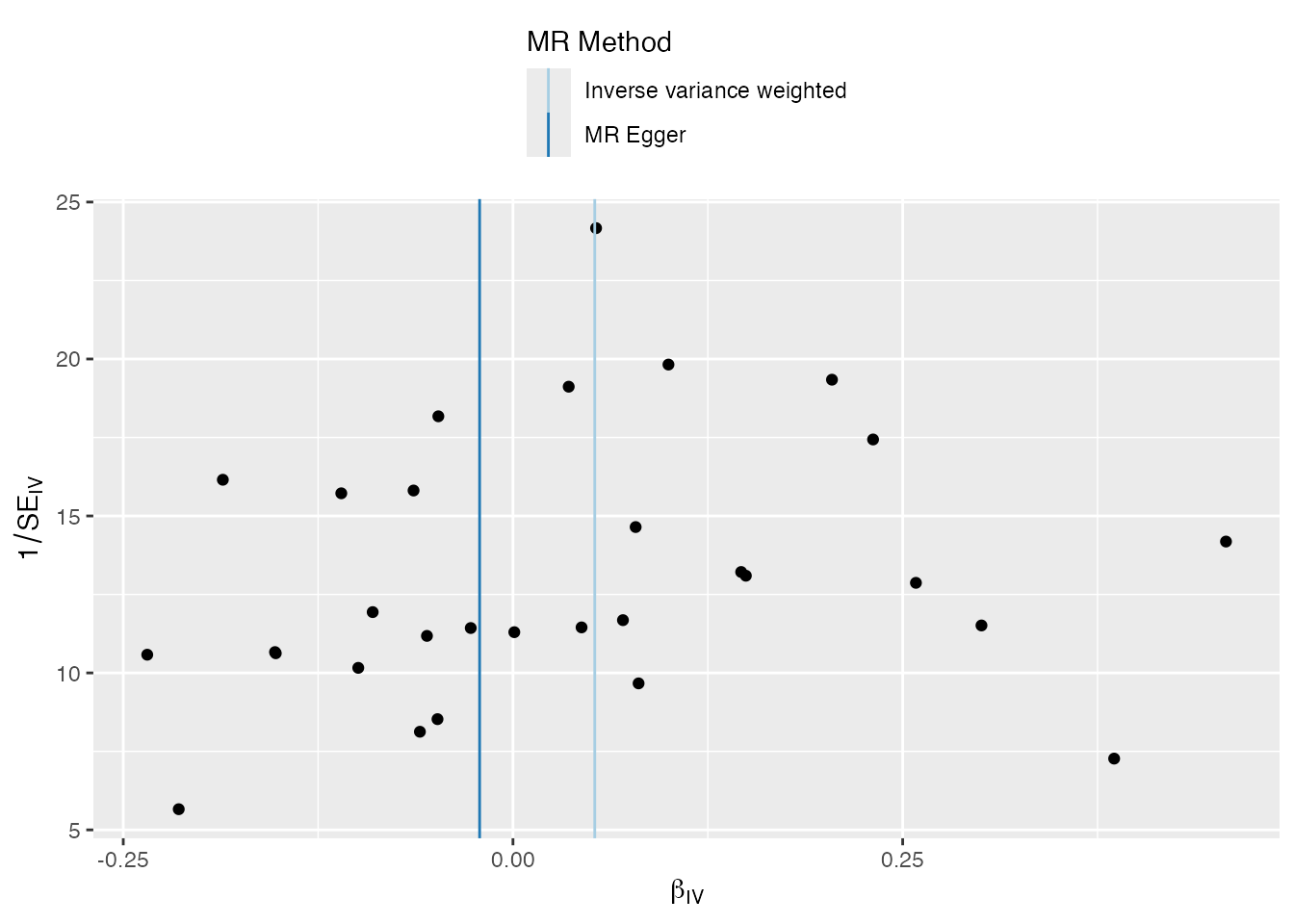
Leave-one-out Plot
Visually identifies SNPs driving the MR result by ordering entries.
loo_res <- TwoSampleMR::mr_leaveoneout(harmonized)
plots <- mr_leaveoneout_plot(loo_res)
plots[[1]]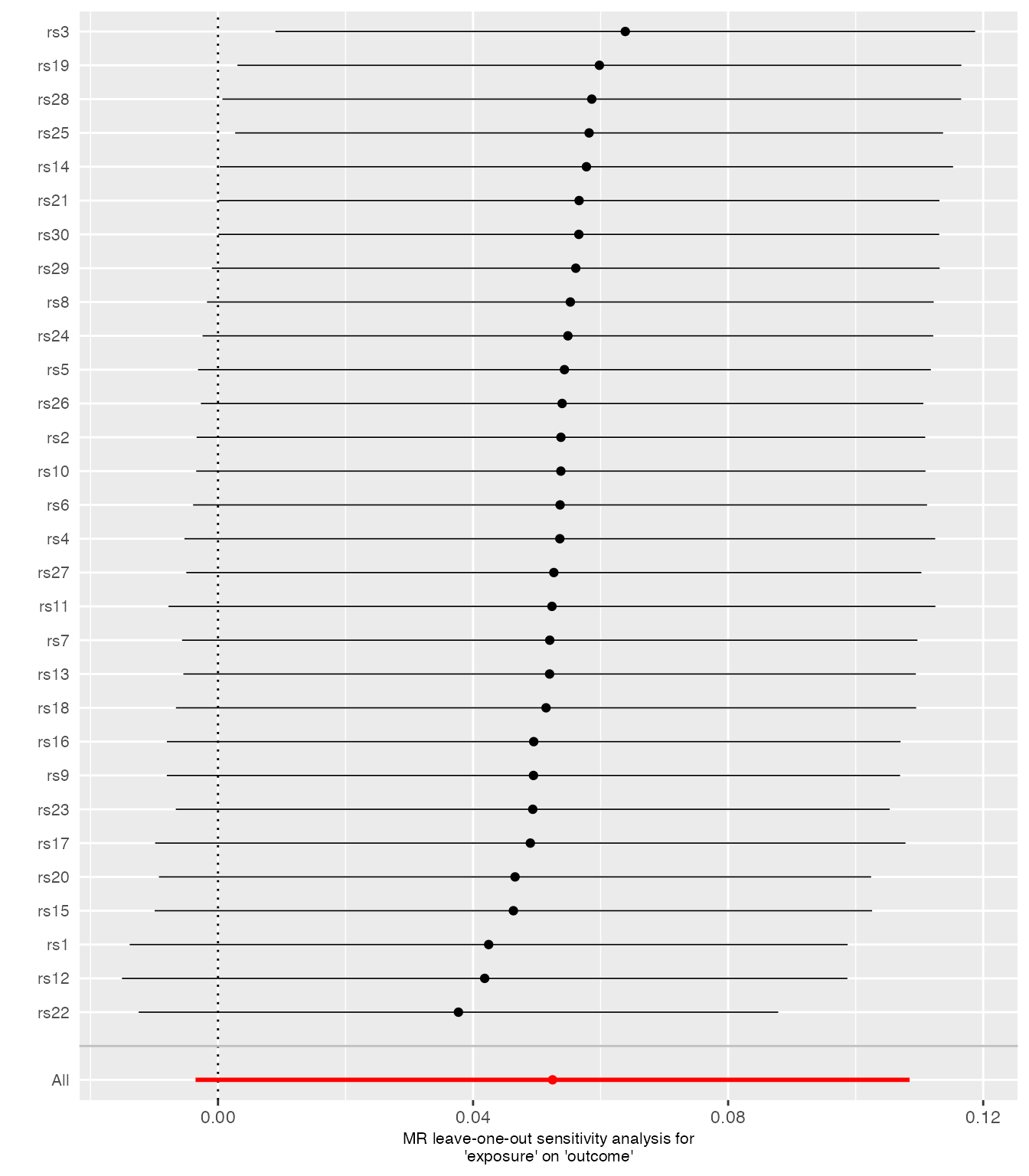
Density Plot
Weighted density estimation of per-SNP effects to visualize the distribution of estimates.
plots <- mr_density_plot(singlesnp_res, res)
plots[[1]]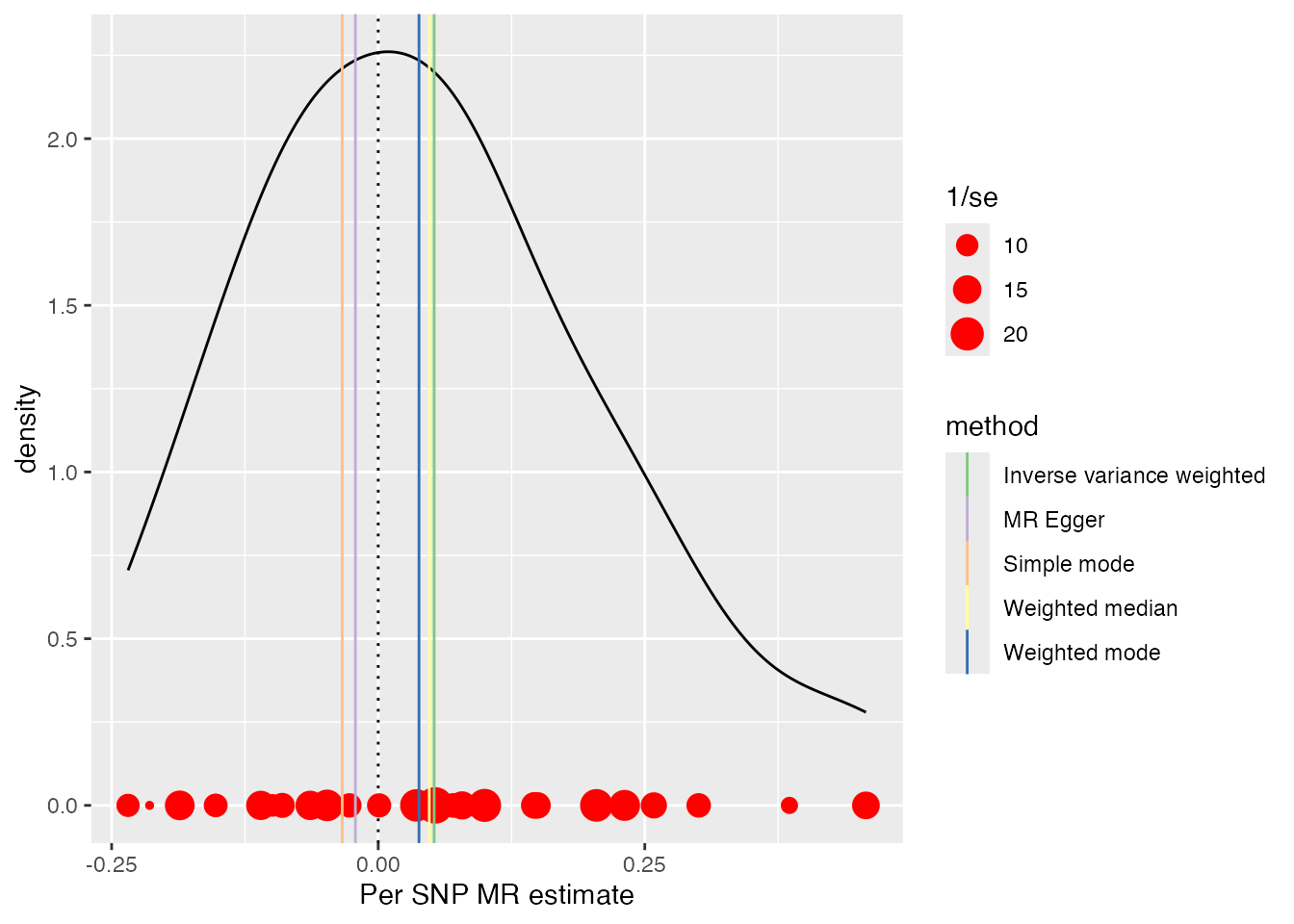
5. Directional Consistency
directional_consistency checks whether the effect
estimate remains in the same direction across different MR results.
# Create 4 results with positive effects for demonstration
set.seed(42)
res1 <- res |> mutate(id.exposure = "Study A", b = abs(b))
res2 <- res |> mutate(id.exposure = "Study B", b = abs(b) * 1.1)
res3 <- res |> mutate(id.exposure = "Study C", b = abs(b) * 0.9)
res4 <- res |> mutate(id.exposure = "Study D", b = abs(b) * 1.5)
# Pivot to wide format as required by the function
wide_res <- bind_rows(res1, res2, res3, res4) |>
select(method, id.exposure, b) |>
pivot_wider(names_from = id.exposure, values_from = b)
# Check consistency
consistency <- directional_consistency(as.data.frame(wide_res))
consistency
#> direction direction_group
#> MR Egger 1 positive
#> Weighted median 1 positive
#> Inverse variance weighted 1 positive
#> Simple mode 1 positive
#> Weighted mode 1 positive