Colocalisation Analysis
colocalisation.RmdIntroduction to Colocalisation
Colocalisation analysis tests whether two traits share a common causal variant at a genomic locus. This is particularly useful in genetics to determine if:
- A GWAS signal and an eQTL signal are driven by the same variant
- Two different traits share the same genetic architecture at a locus
- A disease risk variant operates through a specific molecular mechanism
The coloc package implements Bayesian methods to
evaluate five hypotheses:
- H0: No association with either trait
- H1: Association with trait 1 only
- H2: Association with trait 2 only
- H3: Association with both traits, but different causal variants
- H4: Association with both traits, shared causal variant (colocalisation)
Colocalisation Data Structure
The coloc package requires data in a specific list
format. Each dataset must contain:
Required Components
-
beta: Effect sizes for each SNP (numeric vector) -
varbeta: Variance of effect sizes (numeric vector) -
snp: SNP identifiers (character vector) -
position: Genomic positions (numeric vector) -
type: Either"quant"(quantitative trait) or"cc"(case-control) -
MAF: Minor allele frequency (numeric vector, optional but recommended) -
N: Sample size (single number or vector) -
LD: Linkage disequilibrium matrix (matrix with SNP names as dimnames)
Example Data Preparation
We’ll use the test data from the coloc package to
demonstrate the workflow:
# Load test data from coloc package
data(coloc_test_data)
# Prepare exposure dataset (quantitative trait, e.g., gene expression)
data_coloc_exposure <- coloc_test_data$D1[c("beta", "varbeta", "snp", "position", "type", "sdY", "LD", "MAF")]
data_coloc_exposure$N <- 10000
data_coloc_exposure$chr <- 1
data_coloc_exposure$pval <- runif(n = length(data_coloc_exposure$snp), min = 5e-200, max = 0.1)
# Prepare outcome dataset (case-control trait, e.g., disease)
data_coloc_outcome <- coloc_test_data$D2[c("beta", "varbeta", "snp", "position", "LD", "MAF")]
data_coloc_outcome$type <- "cc"
data_coloc_outcome$N <- 10000
data_coloc_outcome$chr <- 1
data_coloc_outcome$pval <- runif(n = length(data_coloc_outcome$snp), min = 5e-200, max = 0.1)
# Verify structure
str(data_coloc_exposure, max.level = 1)
#> List of 11
#> $ beta : num [1:500] 0.337 0.211 0.257 0.267 0.247 ...
#> $ varbeta : num [1:500] 0.01634 0.00532 0.00748 0.01339 0.00664 ...
#> $ snp : chr [1:500] "s1" "s2" "s3" "s4" ...
#> $ position: int [1:500] 1 2 3 4 5 6 7 8 9 10 ...
#> $ type : chr "quant"
#> $ sdY : num 1.1
#> $ LD : num [1:500, 1:500] 1 0.365 0.54 0.851 0.463 ...
#> ..- attr(*, "dimnames")=List of 2
#> $ MAF : num [1:500] 0.031 0.166 0.0925 0.0405 0.118 ...
#> $ N : num 10000
#> $ chr : num 1
#> $ pval : num [1:500] 0.00808 0.08343 0.06008 0.01572 0.00074 ...
str(data_coloc_outcome, max.level = 1)
#> List of 10
#> $ beta : num [1:500] 0.0926 0.1661 0.1519 0.0342 0.214 ...
#> $ varbeta : num [1:500] 0.01403 0.00467 0.00649 0.01151 0.00593 ...
#> $ snp : chr [1:500] "s1" "s2" "s3" "s4" ...
#> $ position: int [1:500] 1 2 3 4 5 6 7 8 9 10 ...
#> $ LD : num [1:500, 1:500] 1 0.365 0.54 0.851 0.463 ...
#> ..- attr(*, "dimnames")=List of 2
#> $ MAF : num [1:500] 0.031 0.166 0.0925 0.0405 0.118 ...
#> $ type : chr "cc"
#> $ N : num 10000
#> $ chr : num 1
#> $ pval : num [1:500] 0.02032 0.00683 0.03073 0.09931 0.01163 ...Data Quality Checks
Before running colocalisation analysis, always verify your data:
# Check datasets using coloc's built-in validation
coloc::check_dataset(d = data_coloc_exposure, suffix = 1, warn.minp = 5e-8)
#> NULL
coloc::check_dataset(d = data_coloc_outcome, suffix = 2, warn.minp = 5e-8)
#> NULLChecking Data Alignment
The coloc_check_alignment() function verifies that
effect alleles are correctly aligned between datasets by comparing
Z-score products to LD patterns.
# Create alignment plots for both datasets
plot_grid(
coloc_check_alignment(D = data_coloc_exposure),
coloc_check_alignment(D = data_coloc_outcome),
ncol = 2,
labels = c("Exposure", "Outcome")
)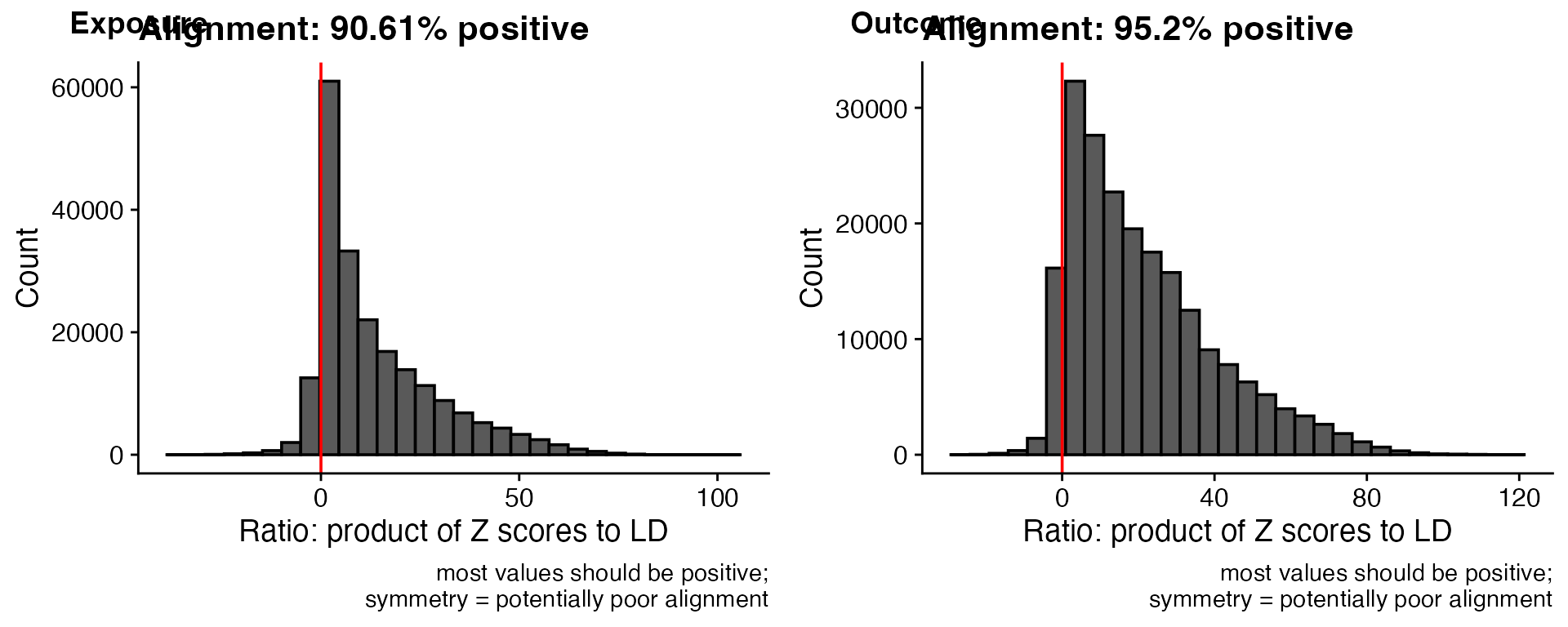
Interpretation: - Most values positive: Good alignment (effect alleles correctly coded) - Symmetric distribution: Poor alignment (potential strand/allele flips) - Aim for >90% positive values for reliable colocalisation results
# Get numeric alignment scores
alignment_exposure <- coloc_check_alignment(data_coloc_exposure, do_plot = FALSE)
alignment_outcome <- coloc_check_alignment(data_coloc_outcome, do_plot = FALSE)
cat("Exposure alignment:", round(alignment_exposure * 100, 2), "% positive\n")
#> Exposure alignment: 90.61 % positive
cat("Outcome alignment:", round(alignment_outcome * 100, 2), "% positive\n")
#> Outcome alignment: 95.2 % positiveVisualizing Datasets
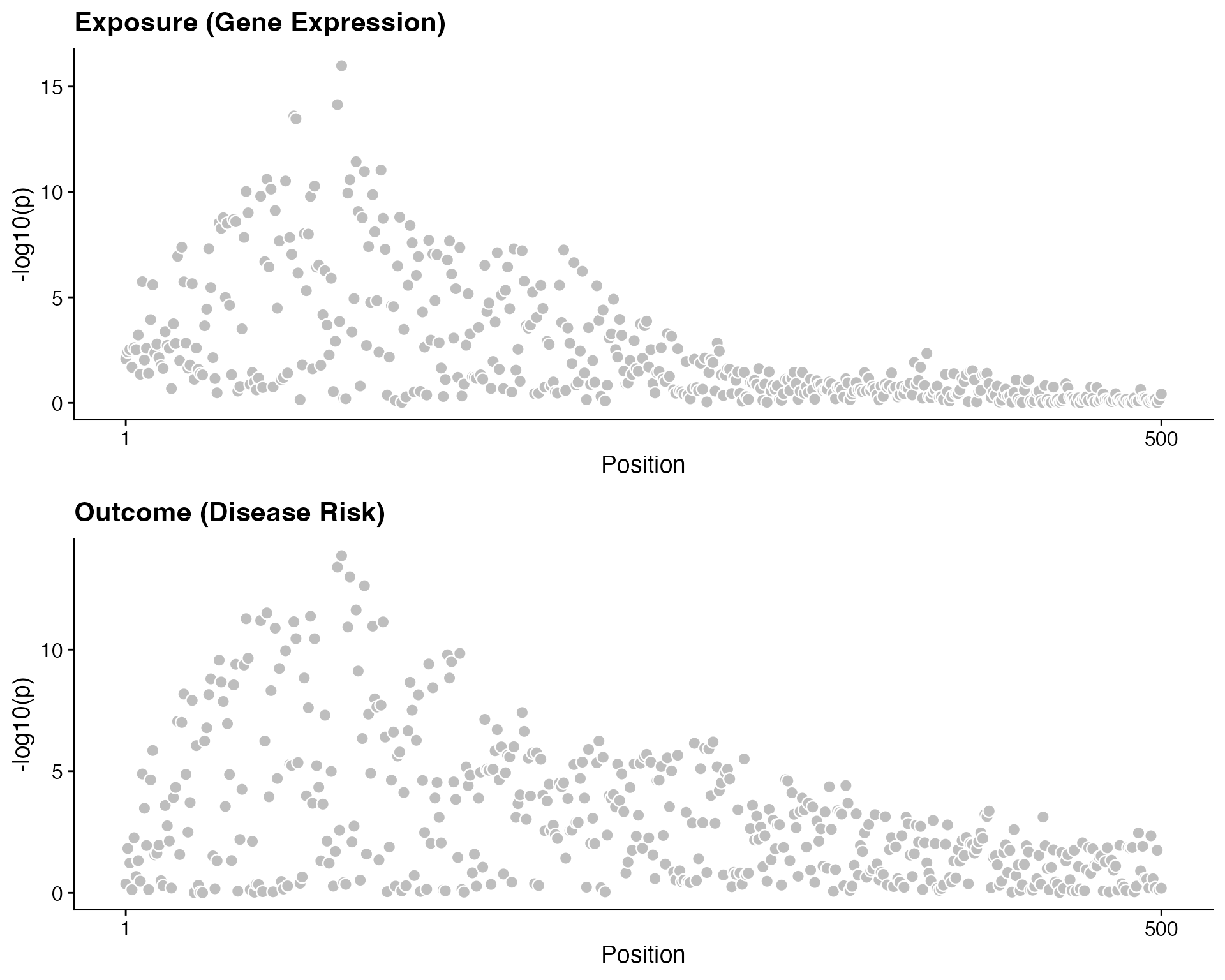
Before colocalisation, visualize the association patterns:
plot_grid(
coloc_plot_dataset(d = data_coloc_exposure, label = "Exposure (Gene Expression)"),
coloc_plot_dataset(d = data_coloc_outcome, label = "Outcome (Disease Risk)"),
ncol = 1
)
Fine-mapping to Identify Causal Variants
Use Bayesian fine-mapping to identify the most likely causal variant in each dataset:
# Fine-map exposure
SNP_causal_exposure <- coloc::finemap.abf(dataset = data_coloc_exposure) |>
dplyr::filter(SNP.PP == max(SNP.PP)) |>
dplyr::select(snp, SNP.PP)
# Fine-map outcome
SNP_causal_outcome <- coloc::finemap.abf(dataset = data_coloc_outcome) |>
dplyr::filter(SNP.PP == max(SNP.PP)) |>
dplyr::select(snp, SNP.PP)
cat(
"Most likely causal SNP in exposure:", SNP_causal_exposure$snp,
"(PP =", round(SNP_causal_exposure$SNP.PP, 3), ")\n"
)
#> Most likely causal SNP in exposure: s105 (PP = 0.956 )
cat(
"Most likely causal SNP in outcome:", SNP_causal_outcome$snp,
"(PP =", round(SNP_causal_outcome$SNP.PP, 3), ")\n"
)
#> Most likely causal SNP in outcome: s105 (PP = 0.592 )Running Colocalisation Analysis
Single Analysis with Default Priors
# Run coloc with default priors
coloc_result <- coloc::coloc.abf(
dataset1 = data_coloc_exposure,
dataset2 = data_coloc_outcome
)
#> PP.H0.abf PP.H1.abf PP.H2.abf PP.H3.abf PP.H4.abf
#> 3.35e-19 7.15e-11 8.19e-12 7.47e-04 9.99e-01
#> [1] "PP abf for shared variant: 99.9%"
# View summary
print(coloc_result$summary)
#> nsnps PP.H0.abf PP.H1.abf PP.H2.abf PP.H3.abf PP.H4.abf
#> 5.000000e+02 3.349762e-19 7.145521e-11 8.188561e-12 7.474844e-04 9.992525e-01Interpreting Results
The posterior probabilities indicate support for each hypothesis:
- PP.H0.abf: No association
- PP.H1.abf: Exposure only
- PP.H2.abf: Outcome only
- PP.H3.abf: Both traits, different variants
- PP.H4.abf: Colocalisation (shared variant)
Decision rules: - PP.H4 > 0.8: Strong evidence for colocalisation - PP.H3 > 0.8: Strong evidence for distinct causal variants - PP.H4 + PP.H3 < 0.8: Inconclusive
Testing Multiple Prior Scenarios
Colocalisation results can be sensitive to prior assumptions. Test multiple scenarios:
# Define different prior scenarios
priors <- list(
list(p1 = 1e-4, p2 = 1e-4, p12 = 1e-5),
list(p1 = 1e-4, p2 = 1e-4, p12 = 1e-6),
list(p1 = 1e-4, p2 = 1e-5, p12 = 1e-6),
list(p1 = 1e-5, p2 = 1e-4, p12 = 1e-6),
list(p1 = 1e-5, p2 = 1e-5, p12 = 1e-7)
)
label_priors <- c(
"p1=1e-4; p2=1e-4; p12=1e-5",
"p1=1e-4; p2=1e-4; p12=1e-6",
"p1=1e-4; p2=1e-5; p12=1e-6",
"p1=1e-5; p2=1e-4; p12=1e-6",
"p1=1e-5; p2=1e-5; p12=1e-7"
)
# Run coloc for each prior scenario
coloc_results <- lapply(priors, function(params) {
coloc::coloc.abf(
dataset1 = data_coloc_exposure,
dataset2 = data_coloc_outcome,
p1 = params$p1,
p2 = params$p2,
p12 = params$p12
)
})
#> PP.H0.abf PP.H1.abf PP.H2.abf PP.H3.abf PP.H4.abf
#> 3.35e-19 7.15e-11 8.19e-12 7.47e-04 9.99e-01
#> [1] "PP abf for shared variant: 99.9%"
#> PP.H0.abf PP.H1.abf PP.H2.abf PP.H3.abf PP.H4.abf
#> 3.33e-18 7.10e-10 8.13e-11 7.42e-03 9.93e-01
#> [1] "PP abf for shared variant: 99.3%"
#> PP.H0.abf PP.H1.abf PP.H2.abf PP.H3.abf PP.H4.abf
#> 3.35e-18 7.15e-10 8.19e-12 7.47e-04 9.99e-01
#> [1] "PP abf for shared variant: 99.9%"
#> PP.H0.abf PP.H1.abf PP.H2.abf PP.H3.abf PP.H4.abf
#> 3.35e-18 7.15e-11 8.19e-11 7.47e-04 9.99e-01
#> [1] "PP abf for shared variant: 99.9%"
#> PP.H0.abf PP.H1.abf PP.H2.abf PP.H3.abf PP.H4.abf
#> 3.35e-17 7.15e-10 8.19e-11 7.47e-04 9.99e-01
#> [1] "PP abf for shared variant: 99.9%"
# Create results table
table_coloc <- data.frame()
for (j in seq_along(coloc_results)) {
results_coloc <- coloc_results[[j]]
results <- data.frame(
scenario = label_priors[j],
nsnps = results_coloc$summary[["nsnps"]],
PP.H0 = results_coloc$summary[["PP.H0.abf"]],
PP.H1 = results_coloc$summary[["PP.H1.abf"]],
PP.H2 = results_coloc$summary[["PP.H2.abf"]],
PP.H3 = results_coloc$summary[["PP.H3.abf"]],
PP.H4 = results_coloc$summary[["PP.H4.abf"]],
p1 = results_coloc$priors[["p1"]],
p2 = results_coloc$priors[["p2"]],
p12 = results_coloc$priors[["p12"]]
)
table_coloc <- dplyr::bind_rows(table_coloc, results)
}
# Display results
print(table_coloc)
#> scenario nsnps PP.H0 PP.H1 PP.H2
#> 1 p1=1e-4; p2=1e-4; p12=1e-5 500 3.349762e-19 7.145521e-11 8.188561e-12
#> 2 p1=1e-4; p2=1e-4; p12=1e-6 500 3.327378e-18 7.097771e-10 8.133842e-11
#> 3 p1=1e-4; p2=1e-5; p12=1e-6 500 3.349762e-18 7.145521e-10 8.188561e-12
#> 4 p1=1e-5; p2=1e-4; p12=1e-6 500 3.349762e-18 7.145521e-11 8.188561e-11
#> 5 p1=1e-5; p2=1e-5; p12=1e-7 500 3.349762e-17 7.145521e-10 8.188561e-11
#> PP.H3 PP.H4 p1 p2 p12
#> 1 0.0007474844 0.9992525 1e-04 1e-04 1e-05
#> 2 0.0074248936 0.9925751 1e-04 1e-04 1e-06
#> 3 0.0007474844 0.9992525 1e-04 1e-05 1e-06
#> 4 0.0007474844 0.9992525 1e-05 1e-04 1e-06
#> 5 0.0007474844 0.9992525 1e-05 1e-05 1e-07Comprehensive Sensitivity Analysis
The coloc_sensitivity() function provides a complete
sensitivity analysis with visualizations:
# Run comprehensive sensitivity analysis
sensitivity_plot <- coloc_sensitivity(
obj = coloc_results[[1]],
rule = "H4 > 0.8",
npoints = 100,
row = 1,
suppress_messages = TRUE,
trait1_title = "Gene Expression",
trait2_title = "Disease Risk",
dataset1 = NULL,
dataset2 = NULL,
data_check_trait1 = data_coloc_exposure,
data_check_trait2 = data_coloc_outcome
)
# Display the sensitivity plot
sensitivity_plot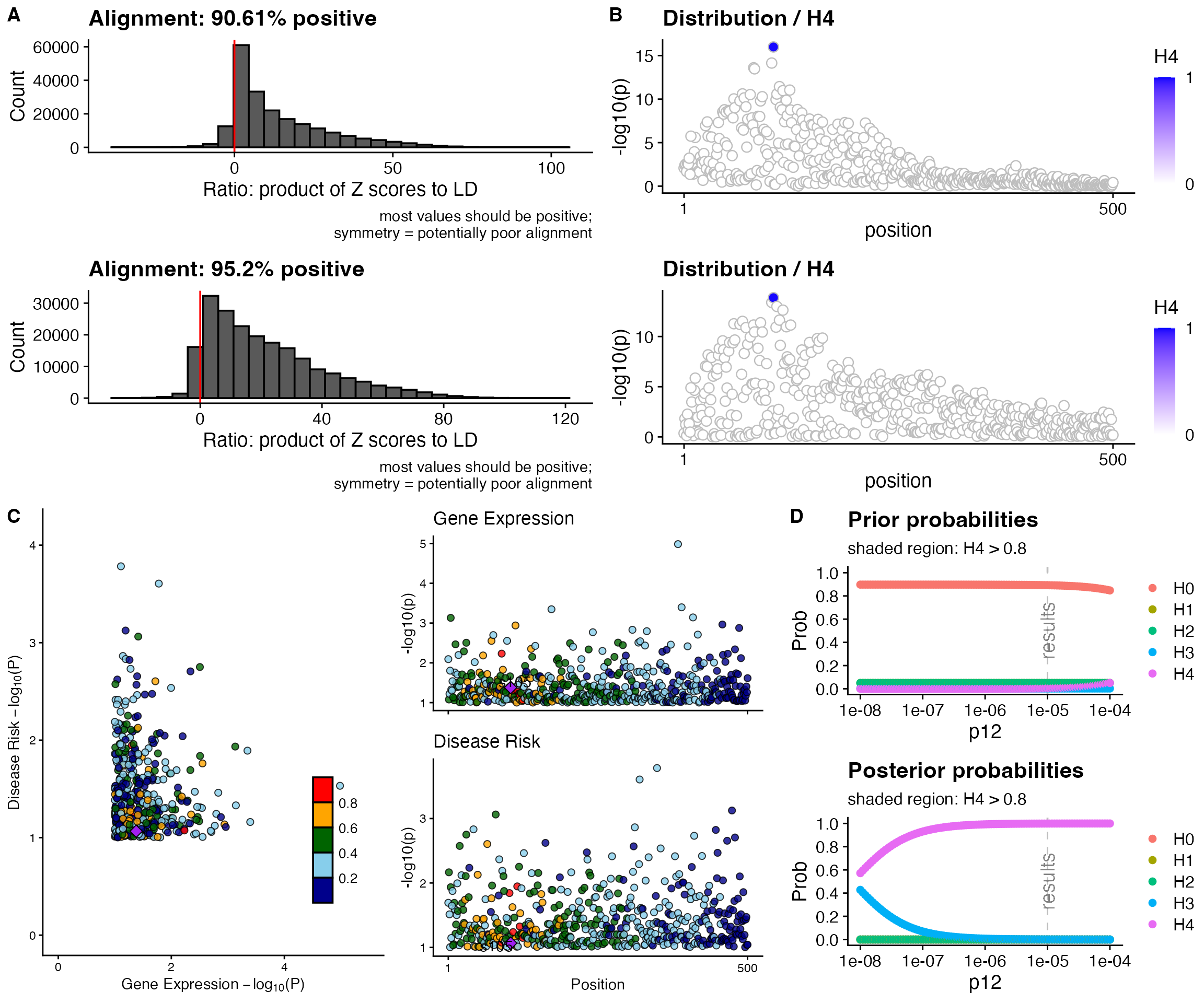
Understanding the Sensitivity Plot
The comprehensive sensitivity plot contains four panels:
Panel A: Alignment Checks - Histograms showing the ratio of Z-score products to LD - Should be predominantly positive (>90%) - Symmetric distributions indicate alignment problems
Panel B: Manhattan Plots - Regional association patterns for both traits - Points colored by SNP-level PP.H4 - Blue indicates higher posterior probability of being the shared causal variant
Panel C: LocusCompare - Scatter plot comparing -log10(p-values) between traits - Points colored by LD with the lead SNP - Helps visualize if the same variant drives both signals
Panel D: Prior/Posterior Sensitivity - Top plot: How prior probabilities change with p12 - Bottom plot: How posterior probabilities change with p12 - Green shaded region: Range of p12 values where PP.H4 > 0.8
Interpreting the green region: - Wide green region: Robust result, insensitive to prior choice ✓ - Narrow green region: Moderately sensitive, interpret with caution - No green region: Weak evidence, highly sensitive to priors
Best Practices
1. Always Check Alignment First
# Poor alignment invalidates results
if (alignment_exposure < 0.7 || alignment_outcome < 0.7) {
warning("Poor alignment detected! Review allele coding before proceeding.")
}Common Issues and Solutions
Issue 1: Low PP.H4 Across All Scenarios
Possible causes: - No true association in one or both traits - Different causal variants (check PP.H3) - Insufficient LD coverage
Solution: - Verify both traits show strong association - Ensure good LD coverage around the signal - Consider expanding the genomic window
Summary
The functions package enhances colocalisation analysis
with:
-
coloc_check_alignment(): Verify data quality before analysis -
coloc_sensitivity(): Comprehensive sensitivity analysis with multi-panel visualizations -
coloc_plot_dataset(): Visualize regional association patterns -
coloc_manh.plot(): Manhattan plots colored by PP.H4 -
coloc_prior.snp2hyp(): Convert SNP-level priors to hypothesis priors -
coloc_prior.adjust(): Adjust posterior probabilities for different priors
Key Takeaways
- Always check alignment before interpreting results
- Test multiple prior scenarios to assess robustness
- Use the sensitivity plot to understand prior dependence
- Report PP.H3 and PP.H4 together for complete interpretation
- Seek replication when results are sensitive to priors
Further Reading
- Giambartolomei et al. (2014). “Bayesian test for colocalisation between pairs of genetic association studies using summary statistics.” PLoS Genetics.
- Wallace (2020). “Eliciting priors and relaxing the single causal variant assumption in colocalisation analyses.” PLoS Genetics.
-
colocpackage documentation: https://chr1swallace.github.io/coloc/